

Tcr/Bcr Receptor Utilities for Solid Tumors

TRUST extracts T/B cell receptor hypervariable CDR3 sequences from unselected tumor RNA-seq data. It is an ultra-sensitive de novo assembly method for calling CDR3s (Li et al., Nature Genetics, 2017), with demonstrated utilities when applied to large cancer genomics data (Li et al., Nature Genetics, 2016). It is written in Python2, with continued updates for performance improvements and additional functionalities. Currently, TRUST supports multiple RNA-seq aligners including Bowtie2, STAR, MapSplice2 etc. It also applies to both hg19 and hg38 human genome references. TRUST can run in parallel mode and uses GPU acceleration to increase computational efficiency. The source code, installation instructions, usage and related dataset are available at: bitbucket.org/liulab/trust/.
Tumor IMmune Estimation Resource

TIMER originally refers to the statistical deconvolution method we developed to estimate six different immune cell types in the tumor microenvironment using gene expression data (Li et al., Genome Biology, 2016). With that work, we published the first TIMER website (cistrome.org/TIMER), and kept a number of useful resources, including tumor purity estimation for over 9,000 TCGA tumors. Later on, we developed an interactive website where users are allowed to perform a number of analysis using the immune infiltration levels we estimated (Li T et al., Cancer Research, 2017), and published the current new website (https://cistrome.shinyapps.io/timer/). Questions can be addressed to Taiwen Li (litaiwen@scu.edu.cn).
Clonal Heterogeneity Analysis Tool
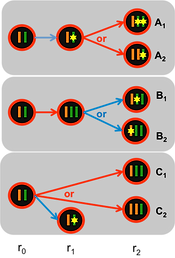
CHAT is a collection of tools for studying intra-tumor heterogeneity using genomics data. The work of CHAT started when we developed a tumor purity inference algorithm (Li et al., Clinical Cancer Research, 2012) and revisited GBM classification. Modifications were made to the framework of this algorithm to allow the inference of the subclonality fractions (sAGP) of tumor somatic copy number alterations. Integrative analysis of sCNA data with whole exome sequencing data enabled the estimation of cancer cell fraction (CCF) of the somatic mutations. The temporal order for a pair of somatic events was also estimated when available (Li et al., Genome Biology, 2014). CHAT is available as an R package, and is also accessible from https://sourceforge.net/projects/clonalhetanalysistool/.
immuno-Similarity Measurement by Aligning Receptors of T cells

iSMART performs a specially parameterized pairwise local alignment on T cell receptor CDR3 sequences to group them into antigen-specific clusters (Li et al., Clinical Cancer Research, 2020). iSMART is developed by Bo Li (bo.li@utsouthwestern.edu), with all rights reserved. iSMART is under GPL3.0 license. iSMART is written in Python2, and is portable without installations. iSMART is accessible from https://github.com/s175573/iSMART.
Deep CNN Model for Cancer Associated TCRs

DeepCAT is a computational method based on convolutional neural network to exclusively identify cancer-associated beta chain TCR hypervariable CDR3 sequences (Li et al., Science Translational Medicine, 2020). The input data were generated from tumor RNA-seq data and TCR repertoire sequencing data of healthy donors. Users do not need to perform training or evaluation. Instead, users can directly apply the PredictCancer function in the package, after downloading the CHKP folder. DeepCat is accessible from https://github.com/s175573/DeepCAT.
Geometric Isometry-based TCR AligNment Algorithm

GIANA (Geometric Isometry-based TCR AligNment Algorithm) is a computationally efficient tool for this task that provides the same level of clustering specificity as TCRdist at 600 times its speed, and without sacrificing accuracy (Zhang, H., Zhan, X. & Li, B. Nat Commun ,2021). GIANA also allows the rapid query of large reference cohorts within minutes. Using GIANA to cluster large-scale TCR datasets provides candidate disease-specific receptors and provides a new solution to repertoire classification. GIANA is accessible from https://github.com/s175573/GIANA.
Mitochondrial-enabled reconstruction of cellular interactions

MERCI (Mitochondrial-enabled reconstruction of cellular interactions) is a statistical deconvolution method for tracing and quantifying mitochondrial trafficking between cancer and T cells. (Zhang, H., Yu, X,. & Li, B. Cancer Cell ,2023).Through rigorous benchmarking and validation, MERCI accurately predicts the recipient cells and their relative mitochondrial compositions. Application of MERCI to human cancer samples identifies a reproducible MT transfer phenotype, with its signature genes involved in cytoskeleton remodeling, energy production, and TNF-α signaling pathways. Moreover, MT transfer is associated with increased cell cycle activity and poor clinical outcome across different cancer types. MERCI is accessible from https://github.com/shyhihihi/MERCI.